
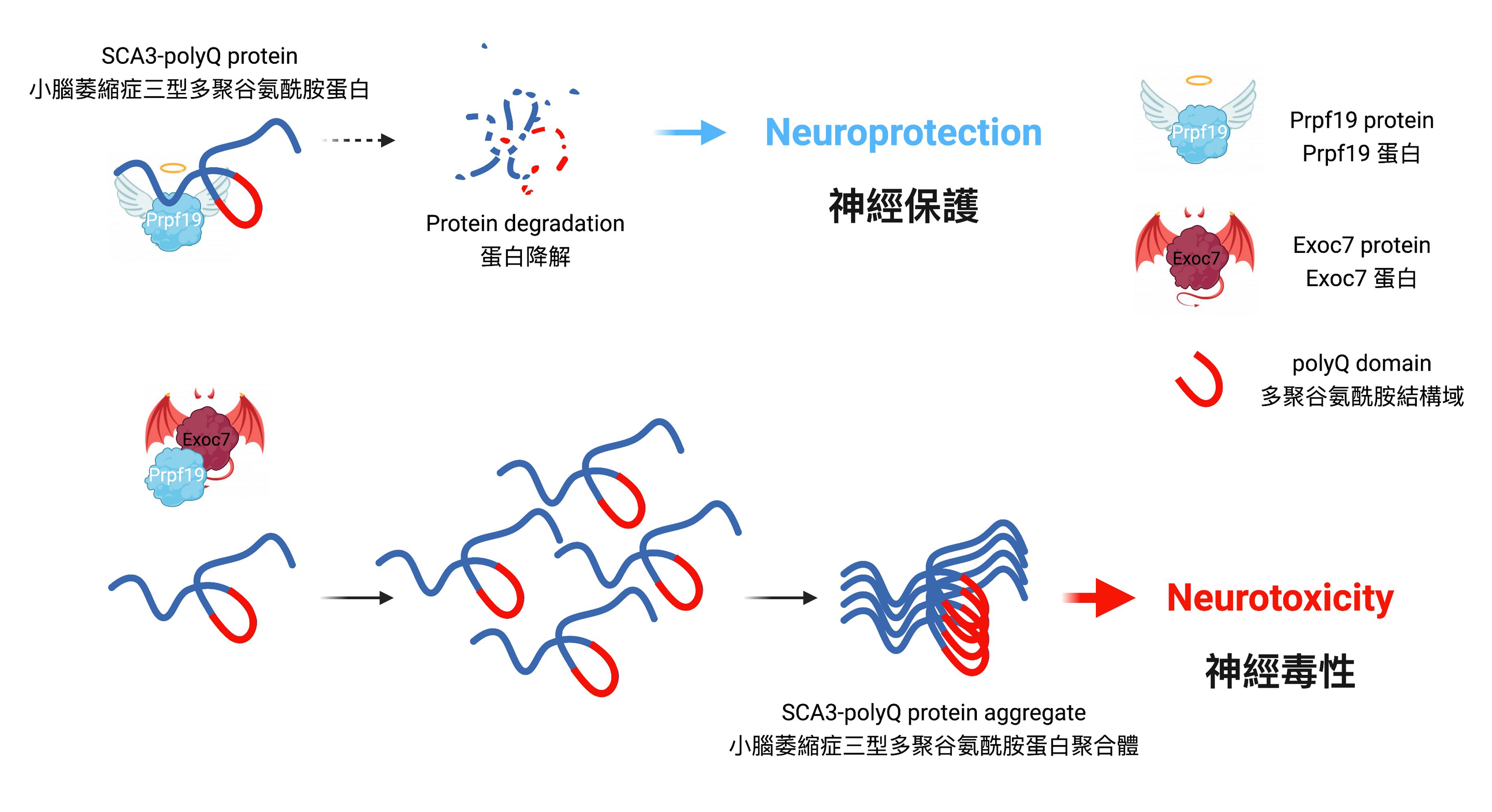
Prpf19 degrades the disease protein of SCA3 and alleviates its toxicity. Exoc7 protein restrains Prpf19 from functioning the degradation, causing it to lose its beneficial effects on SCA3.
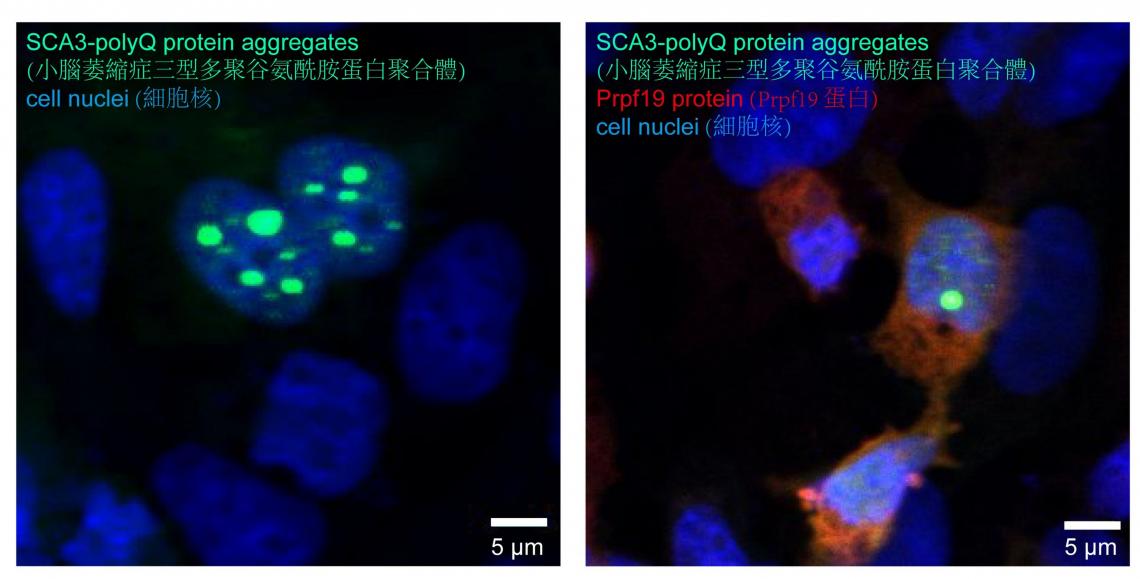
Prpf19 is capable of reducing the level of SCA3-polyQ protein aggregates.
Protein misfolding contributes to the pathogenesis of SCA3
Proteins play a significant role in every single cell development in the human body, including neurons. Numerous studies have proved that misfolds and aggregation of proteins contribute to many occurrences of human diseases. Proteins need to adopt proper folding and architecture before being able to execute their biological functions. Even a minor improper assembly of a protein may result in cellular malfunctioning, leading to toxic insoluble protein aggregates that cause diseases. Progressive misfolding of proteins and aggregates will interfere with the functionalities of other normal proteins, and these are also detected in the deteriorating neurons of SCA3 and other protein misfolding induced disorders, including polyglutamine (polyQ) diseases.
Prpf19 is capable of degrading toxic expanded SCA3-polyQ protein
Professor Edwin Chan, Postdoctoral Fellow Dr. Zhefan Stephen Chen, and the team discovered that the nuclear-localised protein Prpf19 is responsible for scrutinising the quality of SCA3-polyQ, the disease protein of SCA3 or MJD. Potentiating the function of Prpf19 promotes degradation of faculty SCA3-polyQ protein via a process called ubiquitin-proteasome degradation. In this, the toxicity of SCA3 cells is proved to be alleviated, and improvement is also shown in the condition of neurodegeneration and the nervous system of the animal model with SCA3 disease.
The rivalry between Prpf19 and Exoc7 in controlling neurodegeneration of SCA3
Exoc7 is a protein that controls trafficking of proteins within cells, and it is also known as an associating partner of Prpf19. While the coiled-coil domain of Exoc7, a special region of the protein, is crucial for Exoc7 to restrain Prpf19 from functioning, the research team has further discovered that Exoc7 will shuttle to the cell nucleus where it binds directly to Prpf19 and interferes with the pre-mRNA splicing, causing Prpf19 to lose its beneficial effects on SCA3 or MJD disease models.
Professor Chan said, “The current study demonstrates an intricate relationship between Prpf19 and Exoc7, two crucial proteins in nerve cells. Elucidating the mechanism of action of protein networks that govern protein aggregation will allow us to develop potential small molecules or activators targeting Prpf19, with the hope of providing novel strategies for curing SCA3/MJD and other neurodegenerative disorders. Today (28 February) marks the Rare Disease Day 2021 (https://www.rarediseaseday.org/). SCA3/MJD belongs to the category of rare neurodegenerative diseases, I also hope our findings will provide an insight into rare disease translational biomedicine research.”
This work was supported by the General Research Fund and Area of Excellence Scheme of the Research Grants Council of Hong Kong, and The CUHK Gerald Choa Neuroscience Centre. The full text of the research paper can be found: https://www.nature.com/articles/s41419-021-03444-x.
Brief biography of Professor Ho Yin Edwin Chan
Professor Ho Yin Edwin Chan is Professor in the School of Life Sciences, CUHK. He received undergraduate training in biochemistry from CUHK, doctoral training at The University of Cambridge (UK), and postdoctoral training at The University of Pennsylvania (US). Since 1999, Professor Chan has been investigating rare neurological and neuromuscular disorders. In 2014, he established an intercontinental research collaboration network on rare neuronal diseases, including amyotrophic lateral sclerosis/frontotemporal dementia, Huntington’s disease, myotonic dystrophy and spinocerebellar ataxias. Professor Chan actively participates in community services, including as chairperson of the scientific and medical advisory committee of Hong Kong Spinocerebellar Ataxia Association, and Founder of Nexus of Rare Neurodegenerative Diseases. He is a Founding Member and Executive Committee member of The Young Academy of Sciences of Hong Kong.
Brief biography of Dr. Zhefan Stephen Chen
Dr. Zhefan Stephen Chen received his Ph.D. degree at the School of Life Sciences, CUHK. He is currently a Postdoctoral Fellow under the Clinical Neurosciences programme between The Chinese University of Hong Kong and the University of Oxford (Nuffield Department of Clinical Neurosciences and Pembroke College). During his stay in Oxford, Dr. Chen was under the supervision of Professor Kevin Talbot, Professor of Motor Neurone Biology and Head of the Nuffield Department of Clinical Neurosciences, University of Oxford, UK.

