
Transmission electron micrograph of SARS-CoV-2 virus particles, isolated from a patient.
Leading immunologists in Japan are proposing a possible molecular mechanism that causes massive release of proinflammatory cytokines, or a cytokine storm, leading to the acute respiratory distress syndrome (ARDS) in COVID-19 patients. Their suggestions, published in the journal Immunity, are based on recent findings that explain how SARS-CoV-2 enters human cells.
ARDS is a life-threatening condition in which lungs become so inflamed and filled with fluid that they struggle to provide enough oxygen to the body. “To rescue the patients from this condition, it is vital to understand how SARS-CoV-2 triggers the cytokine storm, that leads to ARDS,” explains Masaaki Murakami, the head of immunology laboratory at Hokkaido University’s Institute for Genetic Medicine.
Murakami, together with his collaborator Toshio Hirano from the National Institutes for Quantum and Radiological Science and Technology, reviewed two recent studies by Zhou et al. and Hoffmann et al. in order to understand their implications for finding effective therapeutic strategies for ARDS in COVID-19 patients.
Together, the studies suggest that SARS-CoV-2 enters human cells by attaching to a cell surface receptor called ACE2 and utilizing a human enzyme called TMPRSS2. “Drugs that block the ACE2 receptor or that inhibit the enzyme could help treat the initial stages of the disease,” says Murakami. “However, ARDS with cytokine storm starts to appear in the later phase of infection even when the number of virus decreases. So, there must be another pathway that causes the cytokine storm.”
SARS-CoV-2 is known to be engulfed into the human cell along with the ACE2 receptor it had combined with. “This reduces the number of ACE2 receptors on cells, leading to an increase of a polypeptide, called angiotensin II, in the blood,” says Murakami. Angiotensin II triggers an inflammatory pathway involving NF-κB and IL-6-STAT3 particularly in nonimmune cells including endothelial cells and epithelial cells. “This pathway forms a positive feedback cycle, named IL-6 amplifier, resulting in its excessive activation and therefore the cytokine storm and ARDS,” says Hirano, a pioneer in IL-6 research.
“Part of this pathway involving NF-κB or IL-6-STAT3, or the both, is enhanced with age, which could be why older people are more at-risk of death following COVID-19 infection compared to other age groups,” explains Murakami. “Targeting these pathways, such as with the anti-IL-6 receptor antibody called tocilizumab, could disrupt this life-threatening inflammatory reaction in COVID-19 patients,” Hirano added.
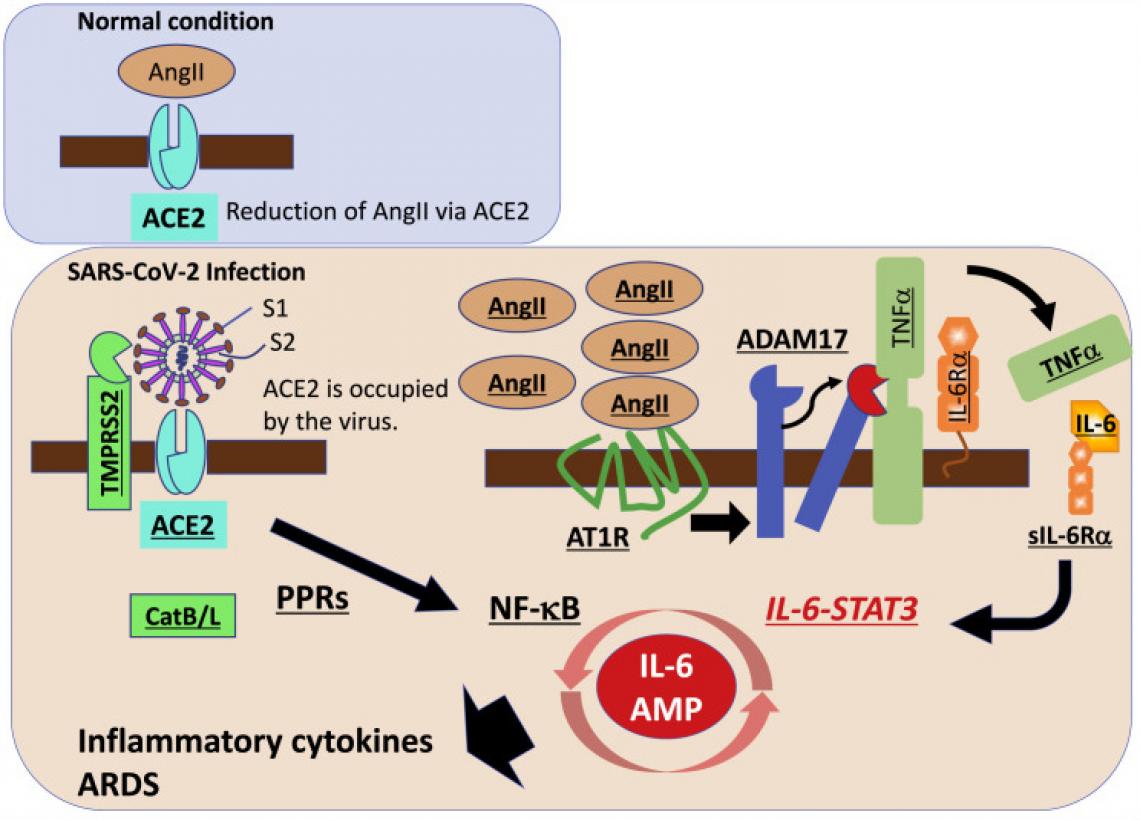
The proposed molecular pathways that lead to the acute respiratory distress syndrome (ARDS) in COVID-19 patients. Drugs targeting to the key molecules such as IL-6 receptor could disrupt the inflammatory reaction.
Contacts:
Professor Masaaki Murakami
Institute for Genetic Medicine
Hokkaido University
Email: murakami[at]igm.hokudai.ac.jp
https://www.igm.hokudai.ac.jp/neuroimmune/Eindex.html
Naoki Namba (Media Officer)
Institute for International Collaboration
Hokkaido University
Tel: +81-11-706-2185
Email: en-press[at]general.hokudai.ac.jp
