
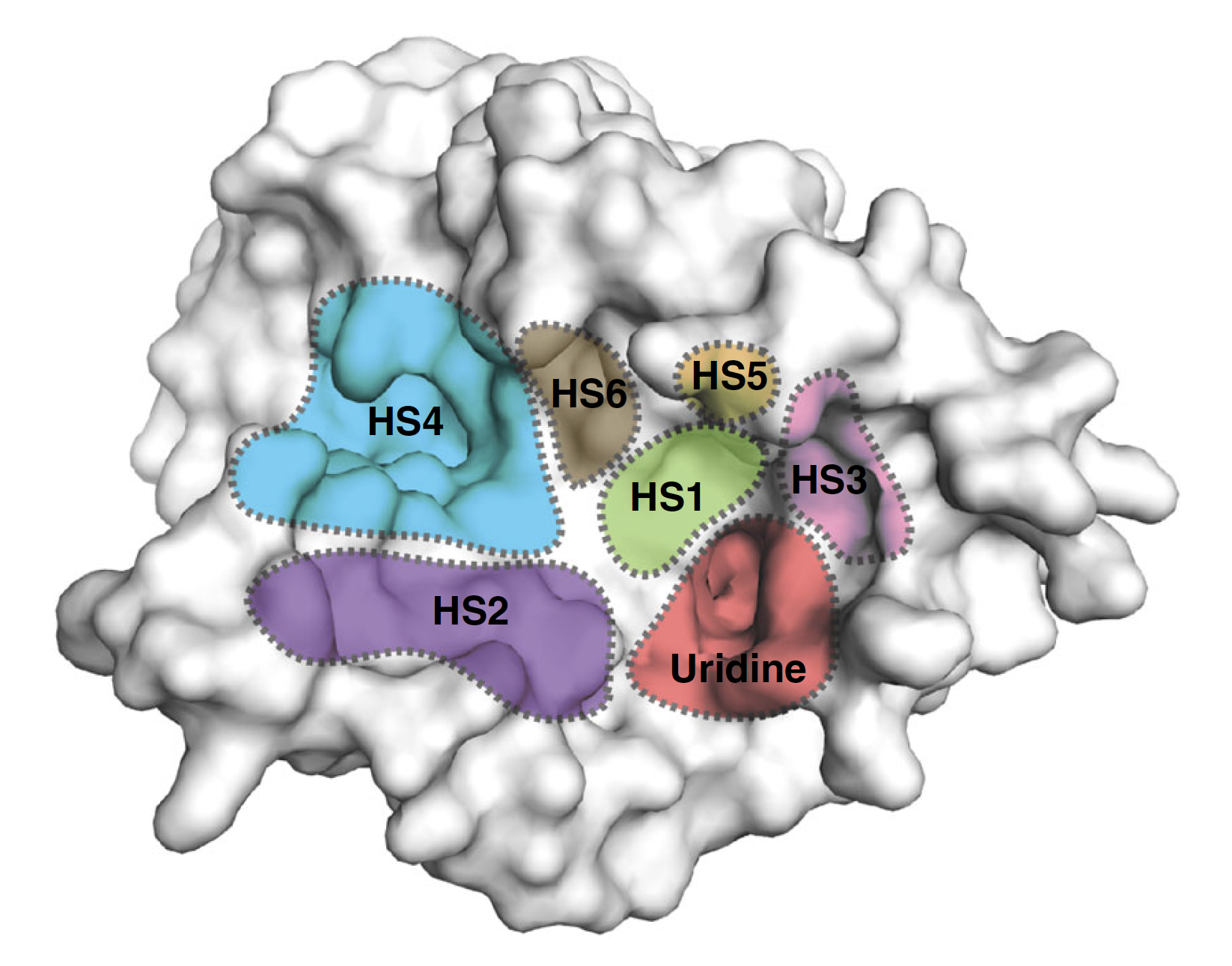
Surface representation of the MraY structure with inhibitor binding sites, or hot spots (HSs), color-coded and labelled as follows: uridine (red), uridine-adjacent (HS1; lime green), TM9b/LoopE (HS2; purple), caprolactam (HS3; pink), hydrophobic (HS4; cyan), Mg2+ (HS5; gold), and tunicamycin (HS6; brown).
Researchers have identified key sections of a bacterial enzyme that could be targeted with new drugs to inhibit the spread of some bacterial infections. A team of scientists from Hokkaido University in Japan, Duke University in the U.S. and The Catholic University of Korea recently published their findings in the journal Nature Communications.
“Drug-resistant bacterial infections have claimed the lives of millions of people worldwide,” said Satoshi Ichikawa of Hokkaido University’s Center for Research and Education on Drug Discovery, a member of the research team. “Because of the emergence of global antibiotic resistance, scientists are constantly searching for new antibacterial compounds.”
One attractive yet unexplored target for new antibiotics is an enzyme called MraY that aids the formation of the bacterial cell wall — an essential component for all bacteria. Five types of natural products have been discovered to have inhibitory activities against MraY. However, exactly what goes on between the inhibitors and MraY on the molecular level has been unclear, hindering drug development.
To get a closer look at these interactions, the researchers analysed the crystal structures of three types of nucleoside inhibitors that target MraY – liposidomycin/caprazamycin, capuramycin, and mureidomycin. Each type of MraY inhibitor has a distinct chemical structure and inhibition mechanism. The scientists used X-ray crystallography, a technique used to determine the atomic and molecular structure of a crystal, to analyse how the inhibitors bind to and interact with MraY.
The structures revealed previously unknown spots on MraY’s surface where inhibitors bind to the enzyme as part of the interference process. Specifically, the researchers found all three types of nucleoside inhibitor bind to one region on MraY’s surface, called the uridine binding pocket, in a similar manner. However, they each form different interactions with at least two other MraY binding sites. These binding sites and pocket could be possible targets for new antibiotics designed to behave like the inhibitors.
“Our work can guide new approaches to MraY-targeted antibiotic design,” says Satoshi Ichikawa. “By using this knowledge of how the compounds bind to and interact with MraY, it would be possible to design an MraY inhibitor that targets a novel combination of binding sites, with improved pharmacological properties and therapeutic potential.”
Contacts:
Professor Satoshi Ichikawa
Faculty of Pharmaceutical Sciences
Hokkaido University
Email: ichikawa[at]pharm.hokudai.ac.jpNaoki Namba (Media Officer)
Institute for International Collaboration
Hokkaido University
Tel: +81-11-706-2185
Email: en-press[at]oia.hokudai.ac.jp
