
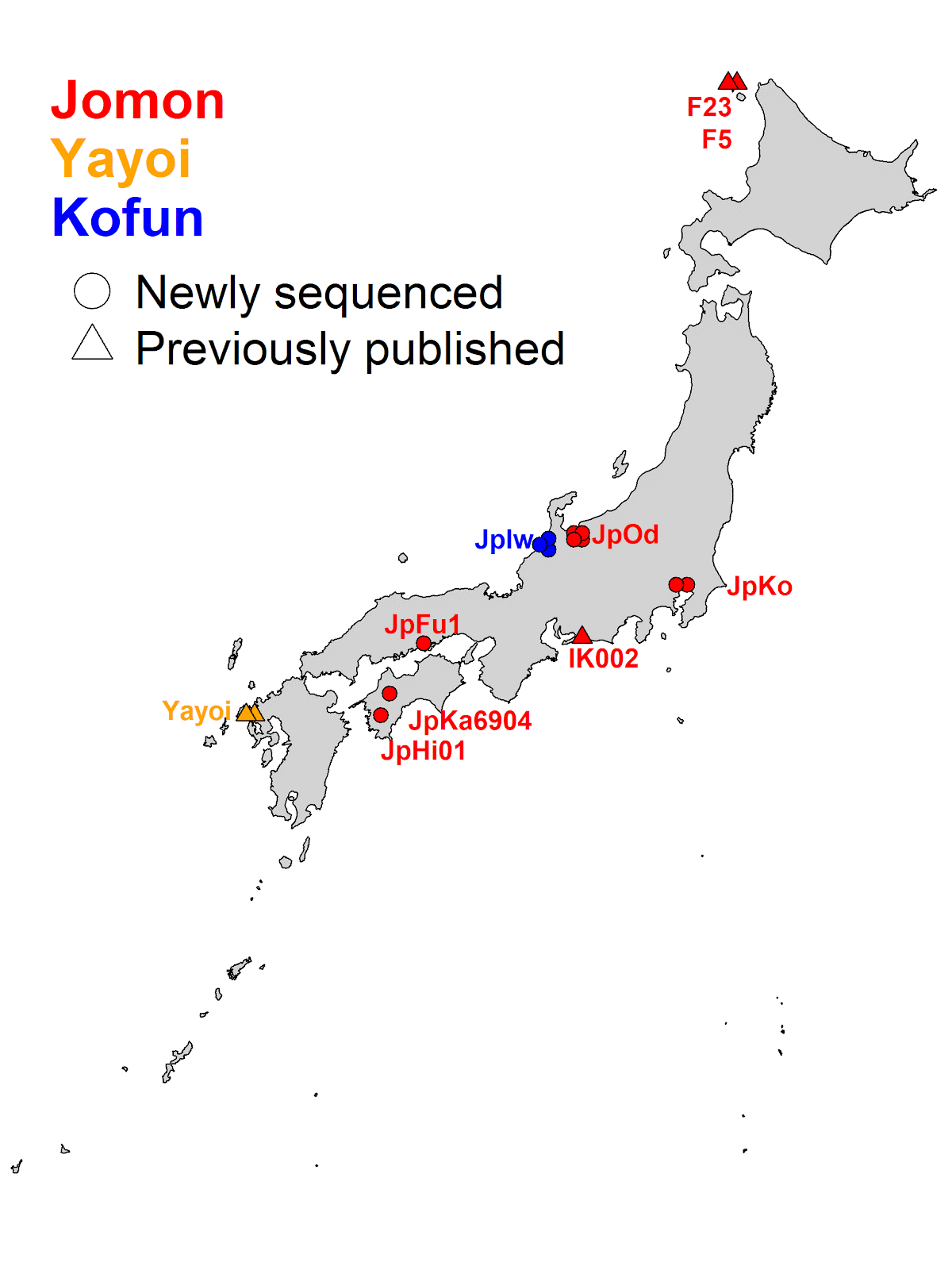
The sites where the samples were newly obtained in this study are indicated by circles and those from previous studies are indicated by triangles. Red, orange and blue symbols indicate the time of the archaeological site, i.e. the Jomon period, Yayoi period and Kofun period, respectively.
Legend:
Jomon sites; IK002 (Ikawazu Kaizuka), F23&F5 (Funadomari Kaizuka), JpKo (Kosaku Kaizuka), JpOd (Odake Kaizuka), JpFu1 (Funagura Kaizuka), JpKa6904 (Kamikuroiwa Iwakage site) and JpHi01 (Hirajo Kaizuka)
Yayoi site; Yayoi (Shimomotoyama Iwakage site)
Kofun site; JpIw (Iwade Yokoana tomb)
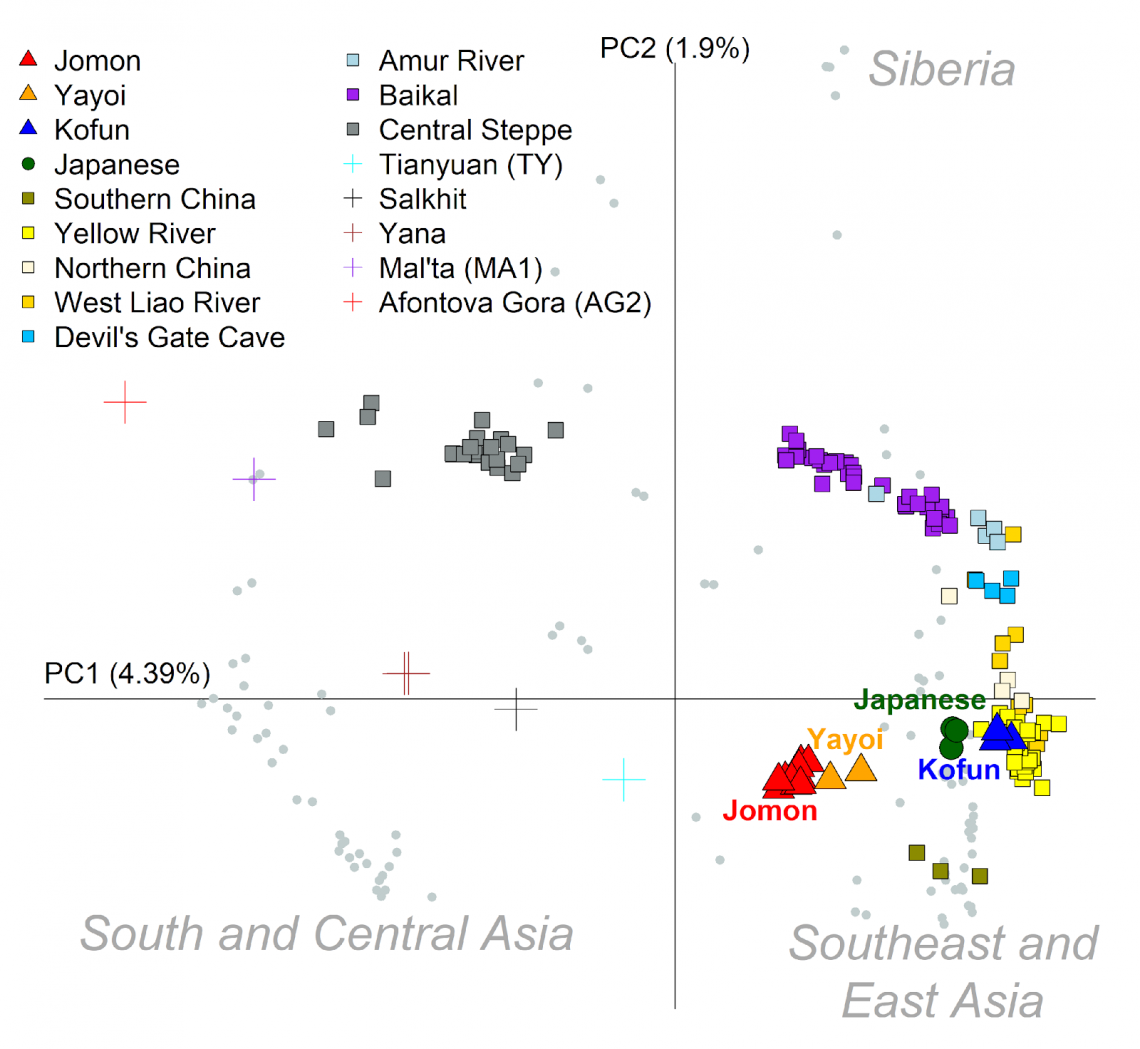
Each symbol indicates an individual, and all modern human populations except the modern Japanese population are shown in gray. Modern populations are roughly divided into three classes: Central and South Asia, Siberia, Southeast and East Asia. The human bones excavated from the archaeological sites are shown in different symbols and colors. For example, the Jomon are indicated by a red triangle, the Yayoi by an orange triangle, and the Kofun by a blue triangle. Crosses indicate individuals of the Paleolithic period. It can be seen that the genetic affinity with the continental population increases with the passage of time from the Jomon.

While the Jomon had a unique ancestral profile, the people having Northeast Asian ancestry in the Yayoi period and those with East Asian ancestry in the Kofun period migrated into the Japanese archipelago and genetic mixture took place. The modern population of the mainland Japan shows the tripartite ancestry structure formed in the Kofun period.
[Background]
Various hypotheses have been proposed in anthropology and archaeology about the origin of the Japanese people. The prehistoric time of Japan is divided into the Jomon, Yayoi and Kofun periods. Thus, elucidation of the origin of prehistoric Japanese populations is important for understanding the relationship between prehistoric people who lived in the Japanese archipelago and modern Japanese. One major hypothesis in physical anthropology is the "dual-structure model"*1) of the Japanese population proposed by Kazuro Hanihara in 1991. This hypothesis expresses the cultural conflict between the Jomon people (hunter-gatherer-fishers) and the Yayoi people (rice farmers) and considers that these two different ancestries have been inherited by the modern Japanese population. However, considering archaeological findings, even in the Kofun period following the Yayoi period, it is now known that major changes occurred in the way of life such as in material culture, e.g. pottery and bronzeware as well as the start of the breeding of horses. The possibility has therefore been suggested that a group of people migrated from a different area of the East Asian continent into the Japanese archipelago during the Kofun period after settlement in Japan during the Yayoi period. A further possibility of human influx from the East Asian continent has been raised even in the Jomon period, which lasted for a long time from about 16,000 years ago to about 3,000 years ago, but the actual situation of such an influx has not been clear.
In recent years, paleogenomics*2) has been used as a powerful tool to elucidate the origins of prehistoric people. By applying paleogenomics, it is possible to obtain the total DNA sequence (human genome), consisting of about 3 billion bases, of humans who once lived in the Japanese archipelago. Thus, the purpose of the present study was to clarify the migration and genetic mixture of human populations that occurred in the transition from the hunter-gatherer-fisher era to the farming era and then to the formation of the national state, by comparing and analyzing the genomic data not only of the Jomon, the Yayoi and the Kofun but also of ancient human bones excavated from archaeological sites on the East Asian continent. With such an approach, we have examined whether the origin of the modern Japanese population could be explained by the conventional "dual-structure model."
[Results]
This study was conducted by an international research team of scientists from Kanazawa University, Trinity College Dublin, Tottori University, Okayama University of Science, Toyama Prefectural Research Office for Archaeological Heritage, Tobinodai Historic Site Park Museum, Ainan Board of Education, and so on. First, the team succeeded in acquiring genomic data of 12 new individuals from ancient human bones excavated from archaeological sites scattered along the Japanese archipelago. In particular, for the Jomon period, comparisons became possible for the first time, not only spanning a wide range of timelines but also at the scale of the whole archipelago. The following six archaeological sites were targets of analyses: the Kamikuroiwa Iwakage Site (rockshelter site) (Kumakogen Town, Ehime Prefecture) from the Initial Jomon period, the Odake Kaizuka (shell mound) (Toyama city, Toyama prefecture) and the Funagura Kaizuka (Kurashiki city, Okayama prefecture) from the Early Jomon period, the Kosaku Kaizuka (Funabashi City, Chiba Prefecture) and the Hirajo Kaizuka (Ainan Town, Ehime Prefecture) from the Late Jomon period, and the Iwade Yokoana Tombs (horizontal tunnel tomb) (Kanazawa City, Ishikawa Prefecture) from the Final Kofun period. The team performed a paleogenomic analysis on the data obtained at these six archaeological sites, together with the human genomes already published in previous studies from the Funatomari Kaizuka (Rebun Island, Hokkaido) and the Ikawazu Kaizuka (Tahara City, Aichi Prefecture) from the Final Jomon period, the Shimomotoyama Iwakage Site (Saseho City, Saga Prefecture) from the Middle Yayoi period and the data from ancient human bones excavated from archaeological sites on the East Asian continent (Fig. 1).
First, the team evaluated the transition of genomic diversity associated with cultural changes in prehistoric times. As a result, it was shown that the genetic affinity to the ancient human population on the East Asian continent became stronger as time progressed from the Jomon period to the Yayoi period and further to the Kofun period. It means that the Yayoi and the Kofun are thought to have ancestry derived from the East Asian continental population. On the other hand, the Jomon were shown to have distinct genetic characteristics from the continental populations. Furthermore, as a result of Model-based Bayesian analysis*3) using the genomic data of the Kamikuroiwa Iwakage Site from the Initial Jomon period, it was shown that the Initial Jomon ancestral population was separated from the continental base population approximately 20,000 to 15,000 years ago and that after separation, they maintained a very small population of ~1000, at least until the Initial Jomon period. It was also shown that during the Yayoi period, when rice cultivation is thought to have been introduced by the migrants, an influx of people from Northeast Asia took place, and that the Yayoi inherited the second ancestry component in addition to that derived from the Jomon. It was further shown that, in addition to these two ancestral components, the Kofun had a third component of East Asian origin so that the cultural transition from the Yayoi to the Kofun period was accompanied by human migration from the East Asian continent and subsequent genetic admixture (Fig. 3). These three components are also found in the genomic sequences of the modern Japanese population (Fig. 3). With these results, this study has thus demonstrated by paleogenomics the "triple structure" of the Japanese genome for the first time.
[Future prospects]
In this study, the team succeeded in acquiring genomic data, at a very high rate, from ancient human bones excavated from archaeological sites representing different eras and regions of the Japanese archipelago. A large number of ancient human bones have been excavated from archaeological sites in the Japanese archipelago. By reusing these ancient human bone materials for paleogenomic research, it is expected that more complex hypotheses will be constructed from the simplified hypotheses proposed so far. It is considered that the "triple structure of the Japanese genome" presented in this study will in the future also be able to more accurately reconstruct the actual process of human migration and genetic admixture within the same era or between different regions by obtaining archipelago-scale paleogenomic data. In addition, there are archaeological sites where the bones of dozens to thousands of individuals are buried. It will therefore be possible to evaluate the kinship and kinship structure by acquiring the genome data of various individuals at a single archaeological site. So far, the kinship and marriage systems of prehistoric people have been inferred indirectly on the basis of objects and archeological events. We will use paleogenomics to reevaluate various current archaeological hypotheses.
[Glossary]
*1) Dual-structure model
An hypothesis about the origin of the Japanese, proposed by Prof. Kazuro Hanihara of the University of Tokyo in 1991, combining research results from natural anthropology and archaeology. This model hypothesizes that the modern Japanese were established by mixture of the indigenous Jomon who had been distributed throughout the Japanese archipelago with the migrants who settled from the East Asian continent during the Yayoi period.
*2) Paleogenomics
Paleogenomics is a research area and method involving the extraction of all genetic information from old biological remains and performing comparative analysis. Paleogenomics allows evaluation of kinship between burials found in archaeological sites based on direct comparisons of the genomic data.
*3) Model-based Bayesian analysis
An analysis method for estimating the original ancestral population number (effective population size) and the time of isolation from other regional populations (branching time), using the genomic data of the population to be analyzed and other regional populations.
